

新药研发到上市流程
本文章收集于网络,如有侵权,请联系,谢谢。
作者:
来源:AIDD Pro
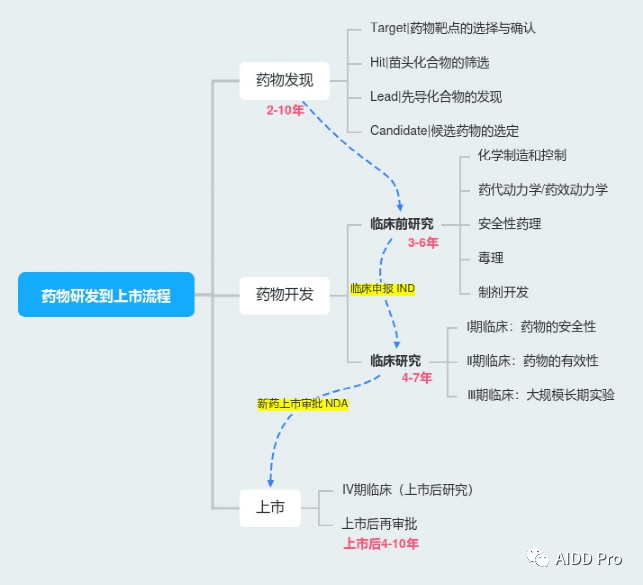
图1 新药研发到上市流程
开发一种新药从最初的想法到最终产品的推出是一个复杂的过程,可能需要12-15年,成本超过10亿美元。下面小编就带大家详细梳理药物从研发到上市的整个流程。
01 药物发现( DrugDiscovery)
1.1 药物靶点(Target)的选择与确认
药物在临床上失败的主要原因有两个:第一是它们不起作用,第二是它们不安全。因此,开发新药最重要的步骤之一就是靶点的选择和确认。
靶点是一个宽泛的术语,它可以适用于一系列生物体,比如蛋白质、基因和RNA。一个好的靶点需要是有效的、安全的、临床可行且有商业价值的,最重要的是,要具有“可药性(druggable)”。所有的药物分子都是通过与靶点结合来引起生物反应的,某些靶点可能更容易结合小分子药物,例如G蛋白偶联受体(GPCRs),而抗体靶点则擅长阻断蛋白质-蛋白质的相互作用。
寻找并确认靶点的方法如图2所示。目前主要应用的有两种:一是利用基因重组技术建立转基因动物模型,或进行基因敲除来验证;二是利用反义寡核苷酸技术(Antisense oligonucleotides,ASOs),通过抑制翻译特定蛋白质的信使 RNA来验证。

图2 靶点选择与确认的方法
1.2 苗头化合物(Hit)的筛选
一旦选定了药物作用的靶点,药物化学家首先要找到对该靶点有作用的化合物。苗头化合物是指对特定靶标或作用环节具有初步活性的化合物。发现苗头物有多种途径,可以从天然产物和化合物库中筛选,也可以进行基于受体或配体结构和机制的分子设计。
根据JMC的研究,最常见的寻找苗头化合物的策略是在先前已知化合物中寻找(43%),其次是随机 高通量筛选(29%),其余的方法包括集中筛选、基于结构的药物设计(SBDD)、基于片段的先导化合物生成(FBLG)和DNA编码库筛选(DEL)等。此外, 虚拟筛选,即使用基于计算机的方法在生物结构的基础上发现新配体的过程,也正在制药行业得到越来越广泛的应用。
1.3 先导化合物(Lead)的发现
一旦通过筛选或其他方式获得了大量的苗头化合物,药物发现团队的第一个任务就是确定哪些化合物是最好的研究对象。一般从多个苗头化合物中, 决策出活性最好的一个(或几个)作为先导化合物用于继续深入研究。先导化合物是已经通过多次实验验证、的确具有一定生理药理活性、可用作进一步深入研究的化合物。是后面大量工作的一个起点。这个决策的过程也被称作“ Hit to Lead”。
这一阶段要对化合物进行更详细的ADME特性分析、细胞毒性测试等,表1显示了要考虑的分析类型。还必须考虑该化合物大规模合成的可行性,以及其GMP制造工艺。

表1 Hit to Lead中要考虑的分析类型
1.4 候选药物(Candidate)的选定
在该阶段,也称为 先导优化(Lead Optimization),生物学家和药物化学家一起努力使先导化合物更安全更有效。一般来说,这一过程通常是反复的,先导化合物需要在遗传毒性模型(如Ames试验)和动物行为学模型(如Irwin试验)中进行检测,得到的活性数据可以结合化合物结构得到初步的 构效关系(SAR)分析,分析结果可以继续指导后续的化合物结构优化。
候选药物(Candidate)就是上面先导优化后找到的“已经很像药的”分子,性质已基本达到期待药物效果。比如如果一种药物是口服的,它应该有足够的溶解度和渗透性,以及可以忽略不计的P-糖蛋白的腔内转运以进行吸收;其次,一旦被吸收,它应该具有与预期用途一致的药代动力学。最后,候选化合物应该有一定的安全性,使风险不会超过预期收益。

图2 候选药物的选定
Hit-Lead-Candidate过程通常需要很长时间,很少有捷径,需要来自不同学科和背景的科学家大量投入。通常对于每个项目,最初可能会筛选20万到超过100万种化合物,在随后的Hit to Lead过程中,会筛选出100种先导化合物,最后优化到1-2个候选化合物。
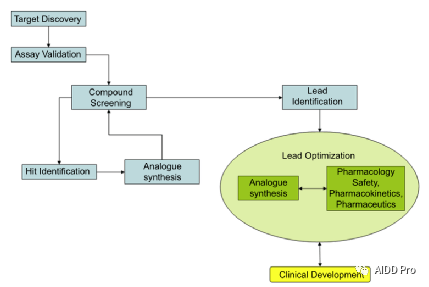
图3 药物发现过程
02 药物开发( DrugDiscovery)
2.1 临床前研究
一旦确定了候选药物,制药公司就会进行临床前安全性、药代动力学、药效学和药剂学等相关研究,以观察化合物针对目标疾病的生物活性,同时对化合物进行安全性评估,以支持临床试验的启动。不同国家和地区启动临床试验所需的具体临床前研究存在一些差异,目前国内主要包括以下几个方面:
- 化学制造和控制(Chemical Manufacture and Control, CMC)
新药开发工作的第一步是原料药合成工艺研发(Process R & D),这是一个不断改进、完善的过程。第一批提供的原料药主要用于毒理研究,要求是越快越好,成本不是主要考虑因素。因此,只要药化路线能够实现毒理批合成,工艺研发部门就会采用。但随着项目的推进,工艺部门会根据需要设计全新合成路线,开发合理生产工艺来满足从I—III期临床用药与商业化的需求;同理,制剂部门首先也会以最简单的形式给药,完成毒理研究,然后不断完成处方工艺研究,开发出商业化的制剂工艺。
- 药代动力学/药效动力学(Pharmacokinetics/ Pharmacodynamic,PK/PD)
了解药物在动物体内的吸收、分布、代谢、排泄(ADME),这些数据可以指导临床研究以何种形式给药(口服、吸入、针剂),给药频率与剂量。
- 安全性药理(Safety Pharmacology)
证明该化合物针对特定目标疾病具有生物活性,同时评估药物对疗效以外的作用,比如可能的副作用,尤其是对心血管、呼吸、中枢神经系统的影响。
- 毒理研究(Toxicology)
毒理研究种类较多,包括急性毒性、亚急性毒性、慢性毒性、生殖毒性、致癌性、致突变性等。为了加速新药能及早验证是否有疗效,尤其是对一些抗癌药,有些耗时费钱的毒理实验(如致癌性、生殖毒性)是可允许在临床试验阶段再进行。
- 制剂开发
制剂开发是药物研发的一个重要环节,随着项目推进,给药方式和处方研究会越来越全面。比如,有的化合物胃肠吸收很差,就需要开发为注射剂;有的化合物在胃酸里会失活,就需要开发为肠溶制剂;有的化合物溶解性不好,也可以通过制剂来部分解决这个问题。
2.2 临床试验申请(Investigational New Application,IND)
在临床前试验完成后,便可向FDA提出IND申请,若FDA在收到后30天内未提出反对意见,申请人便可自行开展新药临床研究[6]。主要目的是提供足够信息来证明药品在人体进行试验是安全的,以及证明针对研究目的的临床方案设计是合理的。大多数经过临床前试验的药物永远不会进入人体试验。
2.3 临床研究
- Ⅰ期临床
如果在几个动物物种中进行了广泛的临床前研究,确定一种化合物是安全的,那么就会在一小部分(一般为20-100名)健康的人类志愿者中启动“首例人体研究”,即Ⅰ期临床。该阶段的主要目标是研究产品的安全性和耐受性,如果可能的话,还会调查其MTD(最大耐受剂量)、药代动力学和药效学。
Ⅰ期临床试验通常要求志愿者住院以进行24小时的密切监护,随着对新药安全性了解的增加,可逐渐提高给药的剂量。
- Ⅱ期临床
Ⅰ期临床研究确定安全的情况下,下一步是在目标疾病患者中(一般为100-500名)测试新药。Ⅱ期临床的结果有助于确定一个药物是否可以科学地和商业地进行到Ⅲ期临床。在不同的时间段采用不同的剂量方案,以获得最好的治疗效果和最佳剂量的建议。
目前,Ⅱ期临床的成功率是药物开发环节中最低的,因为其探索性和患者特异性,药物在患病状态的人体内的作用方式与健康人体内是不同的,对那些影响肠、胃、肝、和肾的药物尤其如此。
- Ⅲ期临床
产品注册前的最后阶段是Ⅲ期临床。在这一阶段,更多患者(一般为1000-5000名)接受更多剂量、更长时间的治疗,其目标是评估药物在长期治疗期间(6个月到1年以上)的安全性。Ⅲ期临床可以说是治疗作用的确证阶段。在大多数情况下,必须完成至少两个设计良好的随机Ⅲ期临床试验,才能向FDA提交新药申请。
在某些情况下,Ⅱ期临床和Ⅲ期临床会合并为一个试验。这些联合试验分几个阶段进行:在早期阶段,确定该化合物是否有足够的研究前景;在后期阶段,将研究药物与对照组进行比较。在适当条件下采用Ⅱ/Ⅲ期综合设计具有样本量小、节省时间和资源、研究持续时间短等优点。
在Ⅰ期临床停止开发的原因通常是由于缺乏具体意义,而在Ⅱ或Ⅲ期终止的原因通常与缺乏疗效有关。
03 上市
3.1 新药上市审批(New Drug Application, NDA)
3.2 Ⅳ期临床(上市后研究)
Ⅳ期临床是为了研究药物在经历上市后更长期治疗后的安全性,并从临床终点的角度确认疗效。这一阶段研究还会涉及到药物配伍使用的研究、药物使用禁忌等。如果批准上市的药物在这一阶段被发现了新的严重不良反应,比如显著增加服药人群心血管疾病发生率,药物会被加注警告说明,甚至下架。
3.3 上市后再审批
药物正式上市销售后,还必须定期向FDA呈交有关资料,包括该药物的副作用情况和质量管理记录。这一阶段的目的是重新审核NDA中的有效性和安全性。
